
Abstract
Idiopathic Pulmonary Fibrosis (IPF) is a debilitating condition, affecting 100,000 Americans with an annual increase of 40,000. This results in decreased respiratory capabilities and the use of supplemental oxygen. Declining lung function in IPF is correlated with the known risk factors of smoking or age over 50. A review of primarily registered clinical trials on IPF is conducted in this article to identify current and future directions of drug development for IPF. Current treatments focus on using the forced vital capacity to improve mortality and other metrics, whereas future therapies are shifting towards slowing or halting disease progression by using agents like GPR84 antagonists, anti-CTGF, and anti-CD20 antibodies. Promising research that warrants further development includes LPA1 receptor antagonist, recombinant human pentraxin 2 biologic, and analog of prostacyclin. However, a recent study did not support treatment with antibiotics such as co-trimoxazole or doxycycline, for the underlying disease. As research progresses, future treatments could be receptor-based because of advances in precision medicine.
Keywords: Idiopathic Pulmonary Fibrosis; Forced Vital Capacity; Extracellular Matrix; Biologics; Antifibrotic agents; LPA1 receptor Antagonists
Introduction
Fibrosis is the overgrowth, stiffening, and/or scarring of tissues that leads to an excess deposition of extracellular matrix (ECM) components. Pulmonary fibrosis (PF) is a chronic progressive lung disease of multiple triggers, in which progression of declining lung function ends in respiratory failure.1, 2 PF is not a single disease. The family of PF diseases is broadly designated as interstitial lung disease (ILD), which includes all diseases of lungs with inflammation and scarring. Over 250,000 individuals in the United States are living with PF and ILD with roughly 50,000 fresh cases being diagnosed each year.3 PF is a spectrum of at least 200 different lung disorders that resemble each other. When the cause of PF is unknown, it is called IPF. IPF is associated with a high incidence of morbidity and mortality. IPF is one of the most common forms of PF, with about 3 million global incidences of IPF being reported in the literature search.2, 4, 5 A systematic review of global incidences of IPF found that North America and Europe have rates between 2.8-9.3 per 100,000 people, with rates being significantly lower in Asia and South America.6 It is estimated that men (20.2/100,000) have slightly higher prevalence than women (13.2/100,000) do.7 Among adults over the age of 65 the prevalence is twice as high. The rising numbers of IPF-related hospital admissions and deaths points to an increasing disease burden.6 IPF has signs that might not present in the early stage outside of dry cough, but as the disease progresses can become problematic when they occur.8 Shortness of breath is a common complaint during exercise and daily activities in patients with IPF. Patients with the IPF may also report fatigue, anxiety, and/or depression.
Different mechanisms have been proposed for the pathogenesis of the IPF. More recent studies suggest that there are more specific and distinctive mechanisms tied to the ECM.9 To understand the impact of the ECM on IPF pathophysiology, an overview of how the lungs work is beneficial. The walls of the air sacs, or alveoli, allow oxygen to enter capillaries and bloodstream in healthy lungs. The lungs are supported by a network of collagen matrices, which are supplied by fibroblasts. Senescent fibroblasts are a type of fibroblasts strongly tied to IPF pathophysiology. They are involved in wound healing and have been identified in the lungs of patients with IPF. Senescence is the state where the cell no longer divides. It is believed to play a key function in both development and wound repair. Typically, once a senescent cell has taken part in wound repair, it is quickly removed from the environment by the immune system. However, if immune clearance cannot remove the senescent fibroblasts, the consistent presence of the senescent fibroblasts is thought to drive disease pathology through their altered secretory profile.10 When a wound occurs, cell division may be necessary. Transforming growth factor-b (TGF-b) regulates a tyrosine kinase pathway involved in cell division. The transcription factor signal transducer and activator of transcription 3 (STAT3) play a role in multiple pathways. These include cell-cycle progression, mitosis, gene transcription, and mitochondrial respiration. All of which are dysregulated during senescence. When TGF-b binds to the tyrosine kinase receptor, the receptor signals internally to the cell to divide, resulting in the production of STAT3. The production of STAT3 results in cell division. Although studies involving STAT3 and IPF are limited, studies show that STAT3 dysregulation is a feature of at least a subtype of patients with IPF.10, 11
The pathophysiology of IPF is thought to be caused by repeated injury to the lungs that prevents normal repair processes from occurring. This can be because of external factors, such as environmental or internal factors, such as genetics. The injury results in activation of the senescent fibroblasts to begin wound repair on the affected lung tissue. However, when the injury is not stopped and becomes continuous, the ability for the immune system to clear the increasing number of senescent fibroblasts may fail. This can cause uncontrolled fibrogenesis.12 This can cause there being more senescent fibroblasts than the body can effectively deal with, resulting in scarring.13 Scarring thickens the alveoli walls in IPF, which makes it difficult to breathe and transport oxygen throughout the body.14 If the injury is not stopped, the lungs can continue to be progressively injured. This can cause structure changes from the collagen tissue production, and oxygen supply capabilities progressively weakening. This results in a steady decline in Forced Vital Capacity (FVC). If unaddressed, the perpetual injury to the lung tissue can cause damage to the alveolar-capillary membrane. When injury continues to outpace repair capabilities, the disruption of the alveolar-capillary membrane allows senescent fibroblasts to deposit collagen tissue in the surrounding ECM, leading to ECM being disrupted.15
IPF may develop in the presence of known risk factors. Various factors that increase the risk of developing PF have been identified, as cigarette smoking, an elderly age, and certain genetic predispositions.3 These risk factors are associated with declining measurements in FVC testing, which measures the amount of air that can be exhaled forcibly from the lungs after inhaling as deeply as possible.16 A retrospective study found that declining FVC measurements over time were associated with higher risk of mortality, respiratory-related hospitalizations, and all causes of hospitalizations.17 IPF presents as a chronic lung condition that is not only is progressive but also is irreversible and inevitably fatal.8 At present, 100,000 people in the United States live with the IPF. Under 200,000 Americans are affected by IPF. Which enables manufacturers to get orphan drug status, for medications approved for IPF.18 Currently, there are a few drugs commercially available in the U.S. drug market and many new molecules and products in the drug pipeline. There are challenges and questions regarding the efficacy, toxicity, and site-specific delivery for FDA approved, as well as pipeline, drugs. This review will focus on registered clinical trials to identify the current status of drug development for the IPF. In this review, some proposed alternative drug delivery approaches for IPF and future directions are also discussed.
IPF is a disease that decreases lung function. It can negatively alter patient quality of life and the lives of caretakers. It can cause the loss of the ability to perform acts of daily living as it affects physical function and self-care abilities. Because of the idiopathic nature, or unknown cause, of IPF, most current treatment approaches, aside from lung transplantation, focus on slowing disease progression but provide no effective treatment of the disease. This approach can still leave the patient with a considerable amount of functional disability. The current standard of treatment is conducted on two fronts: non-pharmacological, which include lifestyle and mechanical or surgical approaches; and pharmacological, which focuses on medications approved to change the pathology of the IPF. The non-pharmacological therapies revolve around oxygen delivery because of the decrease in CO2/O2 exchange capability from the loss of functional lung tissue. These include oxygen therapy, pulmonary rehabilitation, and smoking cessation (if a concern).1 Pharmacological therapies can be broken into two groups: currently approved FDA approaches for IPF and investigational therapies in clinical trials.19-21 Some pharmacotherapy decreases fibroblast activity, under the theory that decreased activity will slow disease progression.22 Steroids, such as prednisone, or immunosuppressive agents, like azathioprine, also operate under this fibroblast theory.10 According to a report prepared by the FDA, many patients have experienced mixed results with the most common forms of therapy, oxygen supplementation, and prednisone. Many claimed to have used over-the-counter (OTC) products prior to being seen for the IPF. Most OTC treatments were an attempt to mitigate symptoms and patients reported mixed experiences with cough medicines.23 A positive effect on respiratory function was observed after three months of supplementation with a combination of vitamins D, C and E in IPF patients. Since routine labs would be expected for patients with IPF, testing for vitamins C, D, and E would be beneficial.24
Method
A literature review was started on clinicaltrials.gov with inclusion criteria of phases 1- 3 with results in the past 15 years. Studies that did not have results available or were not testing pharmacologics in the clinical trial were excluded. Studies where the focus was on an already approved pharmacologic were also excluded and are listed in table 1. The clinical trials that met the above inclusion criteria are listed in a set of tables (Table 2a-c) that highlight different aspects of drugs under evaluation to treat IPF. Table 2. a: Phase 2 and Phase 3 clinical trials that have been shown to have clinical potential, Table 2. b: Clinical trials that have been found to be well tolerated, Table 2. c: Clinical trials that have yet to publish outcomes and Table 2. d: Clinical trials that have been found to not meet the key endpoints.
Results and Discussion
Current Status of IPF Drug Development
The current treatment regimen of IPF has progressed substantially with the addition of two drugs, pirfenidone and nintedanib. These two drugs, pirfenidone and nintedanib, mainly target scar-forming fibroblasts.12 Most treatments including those FVC as reference endpoints. However, these two compounds only slow down the disease progression.25 This will still leave patients with certain amounts of disability, even with treatment. This leaves room for improvement in the treatment options. Two medications are currently approved by the U.S. FDA and are listed in table 1.
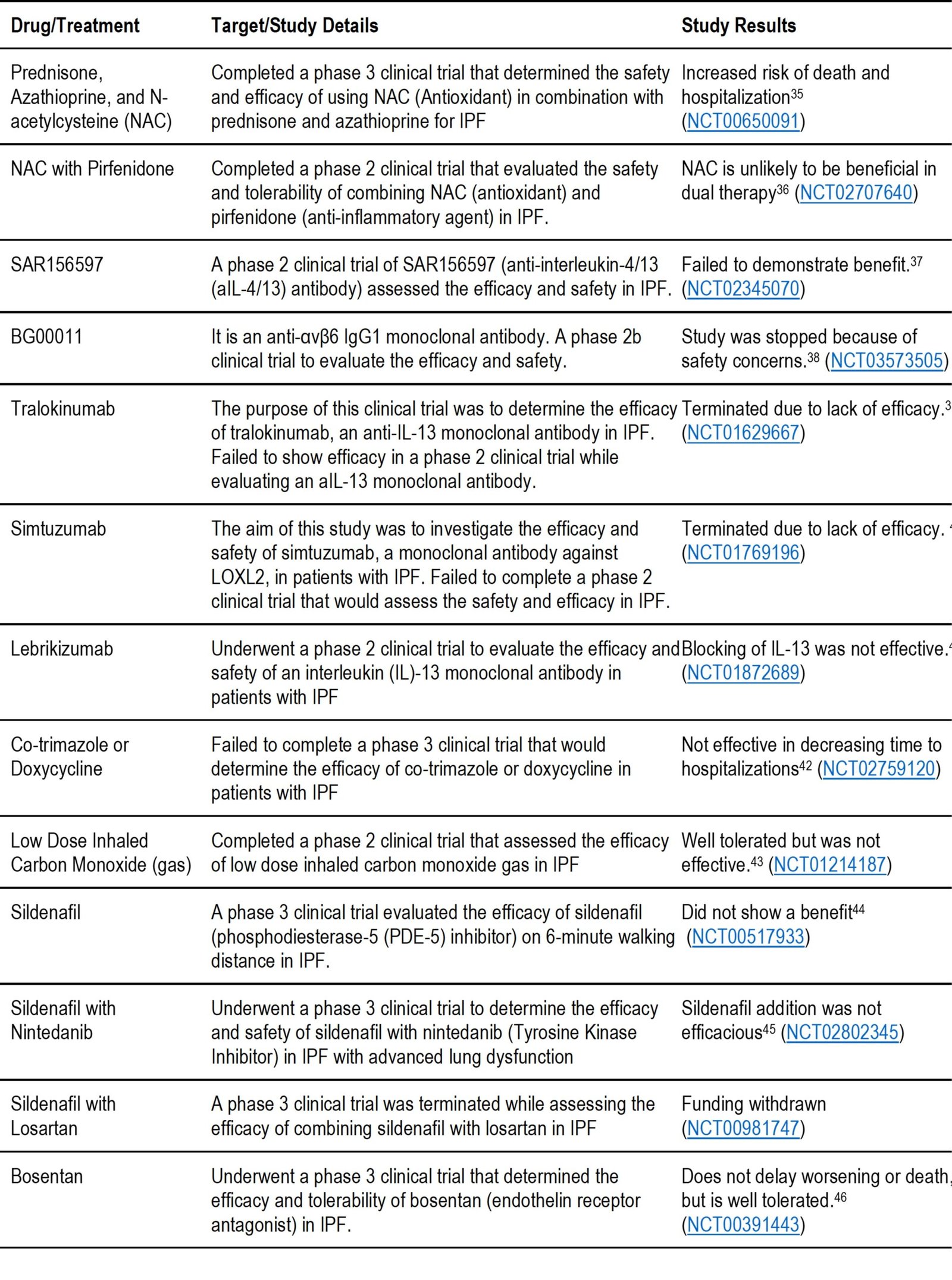
Multiple medications have completed phase 1, 2, & 3 clinical trials and are listed in tables 2a-d depending on their results. These include antifibrotic agents, anti-inflammatory agents, immune cell activators, monoclonal antibodies, autoantibody reductive therapy, stem cells, etc. After reviewing clinical trials for IPF, we have found two drugs, treprostinil and thalidomide, that have completed phase 3 clinical trials. Low dose thalidomide improves quality of life as a cough suppressant, while the inhaled treprostinil has pending results. These are listed in tables 2a for thalidomide and 2d for treprostinil. Clinical trials study results reported in Table 2.d did not meet the key endpoints of the trials. These includes Hemoglobin-Oxygen Affinity Modulator, anticoagulant, phosphodiesterase-5 (PDE-5) inhibitor, anti-interleukin-4/13 (aIL-4/13) antibody, Endothelin Receptor Antagonists, NAC (Antioxidant), some antibiotics, carbon monoxide gas, and some monoclonal antibodies. The authors believe that these pieces of information are still vital for researchers to consider in the early stages of drug design and development.
Table 1. Drugs currently approved by U.S. FDA to treat IPF patients
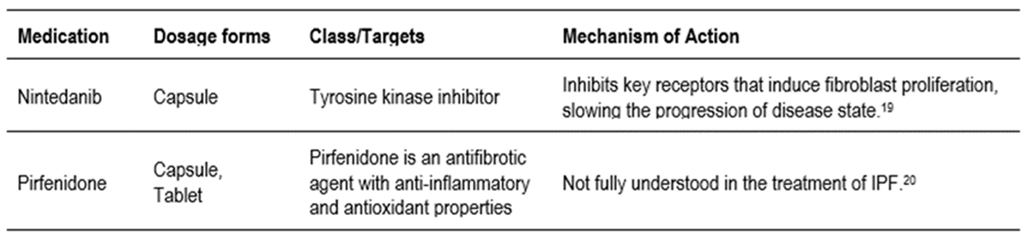
Table 2. a: Phase 2 & 3 clinical trials that have been shown to have clinical potential by meeting key trial endpoints.
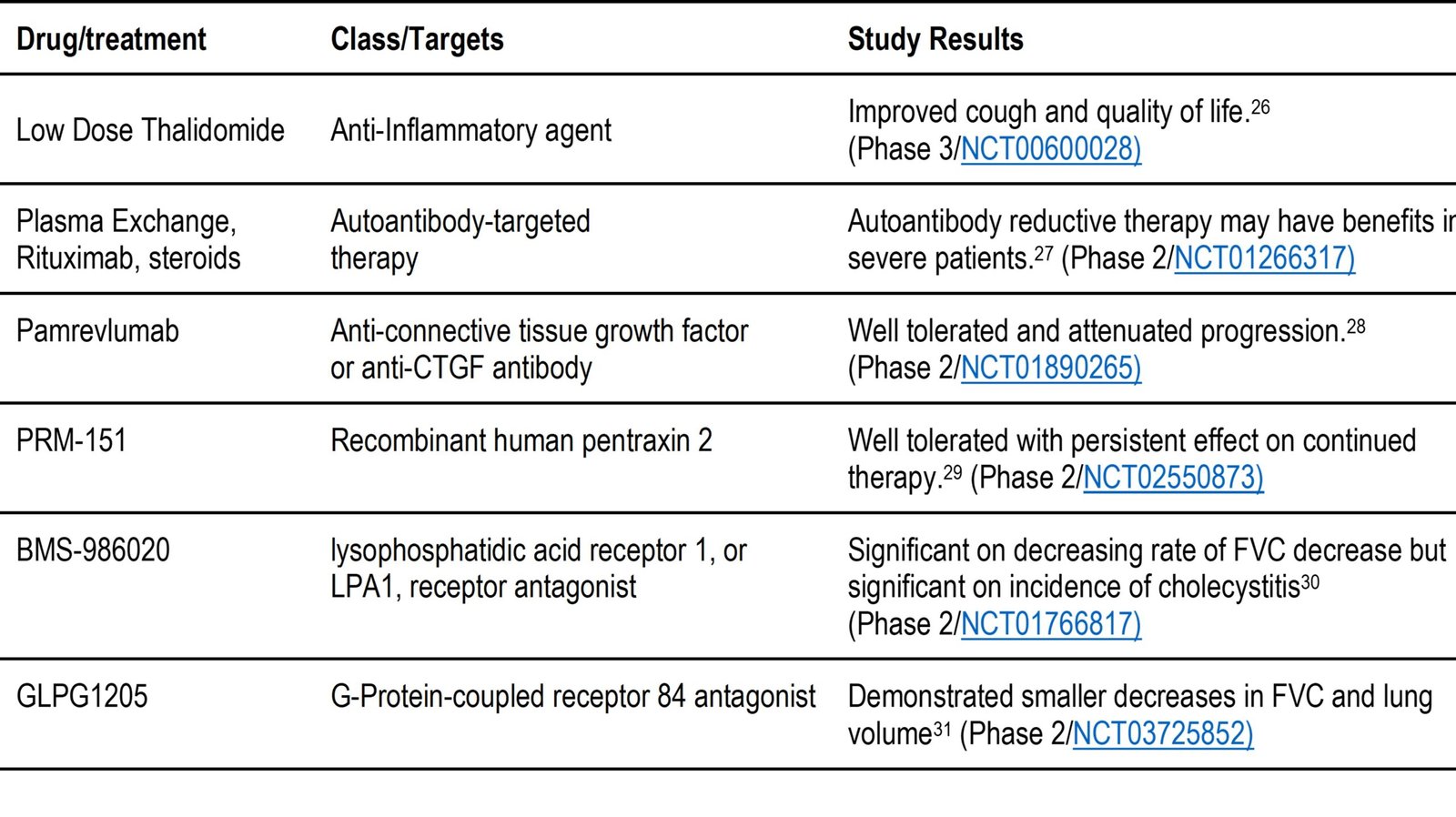
Table 2. b: Drugs under evaluation to treat IPF that have been found to be well tolerated.
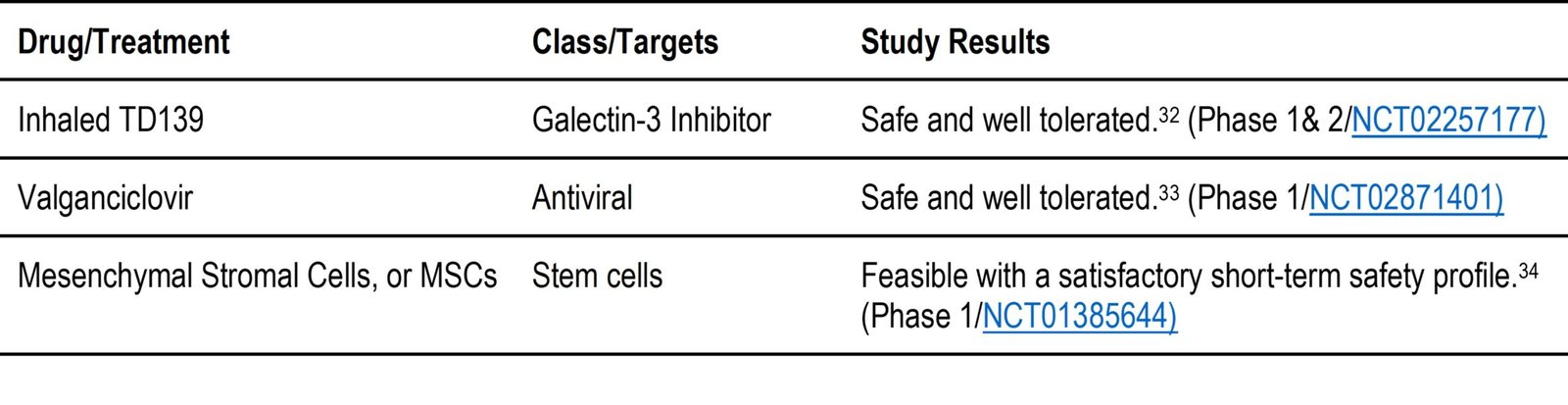
Table 2. c: Drugs under evaluation to treat IPF that have yet to publish outcomes of the clinical trial.
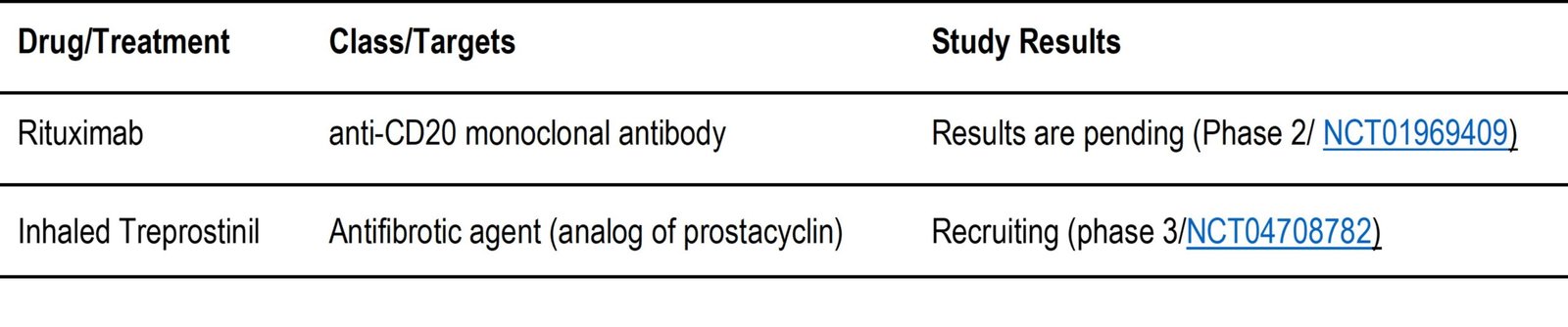
Table 2. d: Medications currently being evaluated in clinical trials that have been found to NOT meet the key endpoints of the trial.
Challenges in for Clinical Drug Development in IPF
Despite decades of research resulting in increased knowledge on IPF, only two medications have been approved by U.S. FDA to treat IPF. Both of the drugs only slow down disease progression. Challenges to developing targeted therapies for IPF are many. They include the unknown etiology of the disease, effective targets, demographics of the underlying patient base, preclinical and clinical studies, and delivery of drug at the site of action (lungs).
- Researchers spend a significant amount of time identifying new therapies during preclinical studies. This is because IPF’s complex pathogenesis involves trying multiple treatments targeting different disease pathways to see what works.
- The progressive nature of IPF makes patient demographics in IPF studies difficult. For example, patients might not complete the study, which can affect its significance.
- All treatment strategies have challenges related to successful drug delivery at the site of action. Delivering drugs directly to the lungs can increase efficacy and reduce toxicity. Several barriers prevent inhaled drugs from reaching the lungs. The first barrier is the structure of the lung and the second barrier is the pulmonary drug delivery devices.
Discussion with future directions
Researcher develops therapies in two categories; either to improve the quality of life or to stop or slow down the progression of the disease. The U.S. FDA has approved two drugs, pirfenidone and nintedanib. Both drugs primarily target scar-forming senescent fibroblasts to slow the progression of the disease. Recent data suggests that the persistent presence of senescent cells could be serious, and their beneficial effects can become dysfunctional early in life, leading to disease later.47 It is therefore important to develop strategies to prevent the accumulation of senescent cells to slow the rate of fibrosis or scarring in the lungs. The clinical trial reports also show that several drugs are in clinical trials targeting scar-forming fibroblasts in the anti-fibrotic agent category. These include pamrevlumab, inhaled treprostinil, PRM-151, BMS-986020, GLPG1205. Below, we discuss the results, goals and future directions falling under antifibrotic agents.
- In the phase 2 study, pamrevlumab slowed the progression of IPF and was well tolerated. It is a fully recombinant human monoclonal antibody (rHMAB) directed against the connective tissue growth factor (CTGF) in IPF. CTGF is a secreted glycoprotein that plays a central role in the process of fibrosis. Pamrevlumab has subsequently received Fast Track Designation and is completing a Phase 3 study under NCT03955146 and is showing promise as a novel, safe and effective treatment for IPF.28
- PRM-151 has advanced to Phase 3 studies under NCT04552899. It is a recombinant human pentraxin-2 protein that reduced PF in preclinical models of TGF-1 overexpression and bleomycin-induced PF; the effect lasted up to 30 days after ingestion. Pentraxin 2 is also a potent inhibitor of monocyte differentiation into pro-inflammatory macrophages and the production of transforming growth factor (TGF)-1. This is a key mediator of PF. Plasma concentrations of pentraxin 2 are reduced in patients with IPF and correlate with disease severity, further supporting its role in modulating fibrosis.29
- BMS-986020 (LPA1 receptor antagonist) proved to be significant in reducing the rate of FVC with the side effect of cholecystitis (gallstones). While the target, LPA1, is promising, we suggest that this can be addressed through surgery. Surgical removal not only addresses the side effect of BMS-986020 for IPF but potentially improves the patient’s quality of life. Patients can also enjoy a good quality of life without the gallbladder, which would address concerns about BMS-986020’s side effects. Modification of the drug during development to reduce the occurrence of gallstones should be considered.30
- Phase 2 clinical trials of the drug GLPG1205 resulted in less reduction in FVC, consistent with a therapeutic effect. The G protein-coupled receptor 84 (GPR84), which is activated by a variety of medium-chain fatty acids, is involved in fibrotic processes. GLPG1205, a selective GPR84 antagonist, inhibits the migration and activation of monocytes and neutrophils. This alone, together with the observed safety profile for GLPG1205, supports further development. The most common adverse events (AEs) for GLPG1205 alone were GI disorders, particularly nausea. No relevant safety signals were seen for GLPG1205 alone or with pirfenidone.31
- TD139 has been evaluated in a phase 1 and 2 combination clinical trial using DPI administration. Galectin-3 inhibition acts as an antifibrotic agent. This has been shown to block recruitment and expansion of Gal-3-secreting macrophages, which drive local myofibroblast activation and slow fibrogenesis. Effects on the sense of smell or taste have been reported with predominantly good tolerability.32
- Inhaled treprostinil has shown antifibrotic effects in preclinical studies. This supports the investigation into the treatment of IPF with inhaled treprostinil as an antifibrotic agent. This is being conducted as a phase 3 clinical trial under NCT04708782 using ultrasonic nebulization to deliver inhaled treprostinil.48
Anti-inflammatory agents are of interest because of the inherent association that fibrogenesis plays a role in active site healing. In a normal inflammatory process, when healing is required, fibroblasts form a temporary bandage of scar tissue that is later removed. The inflammation during this process acts as a signal to bring the components necessary for healing to the site of healing. In IPF, excessive injury to the lung tissue leads to excessive inflammation and thereby excessive fibroblasts in the area. Anti-inflammatory drugs work by reducing the inflammatory signal. This decreases the number of fibroblasts that need to be in the area, thereby potentially reducing the amount of scar tissue. This therapy may be used in combination with other trial drugs to potentially improve efficacy.
- Pirfenidone is a U.S. FDA approved antifibrotic agent with anti-inflammatory and antioxidant properties, commonly used in combination with trial drugs for IPF.
- In a phase 3 clinical trial under NCT00600028, the anti-inflammatory effects of low-dose thalidomide were found to suppress chronic cough in IPF patients.
Biologics are becoming an integral part of the targeted treatment of many diseases because of their effectiveness and the possible targeting of therapy. Biological therapies are being studied to treat patients suffering from various forms of ILD. This includes IPF. Part of the utility of biologics is not just specificity, but how they may affect the ECM. The ECM also plays an important role in IPF. Following its discovery, many investigators are turning their attention more to ECM-focused therapies, using targeted biologics alongside other therapies.15 Because excess fibrotic tissue could disrupt the ECM if left unchecked, exploration of specific targets becomes important to highlight targets other than CTGF. Given that targets such as CTGF and CD20 are effective, this further raises the importance of the ECM in the pathophysiology of future therapies in IPF. This is where gene therapy can play a role by identifying common biomarkers in IPF that could be used to design therapies.49 Biologics being studied in the clinical trials include autoantibody reduction therapy, MSCs, and monoclonal antibodies.
- Reductive therapy with autoantibodies is being studied in phase 2 under NCT01266317 as a combination therapy with rituximab, plasma exchange and steroids. Antibodies are produced by the immune system to fight infection. They do this by distinguishing foreign objects, such as bacteria and viruses, from our own bodies. Patients diagnosed with IPF have antibodies (called autoantibodies) that treat the lungs as foreign objects and attack them, causing scars and injuries. As the disease progresses, many people have elevated levels of autoantibodies in their blood and lungs, which could make the disease worse.50 Antibody-reducing therapy with rituximab would be useful in patients with elevated autoantibodies.
- In a phase 2 clinical trial under NCT02594839, MSCs were found to have dose-dependent efficacy results. MSCs are a type of stem cells found predominantly in lungs that are IPF deficient, and MSC therapy attempts to replace such a deficiency. As a type of stem cell, they try to stimulate repair of injured lung tissue and have been found to be well tolerated in previous studies. High-dose MSCs were found to be more effective than low-dose MSCs in lung diseases, such as IPF.51
- Monoclonal antibodies (MABs) are being investigated in various studies with mixed results.This is because MABs are designed to specifically block a unique target. The results of whether the target is beneficial for inhibiting a disease are usually revealed in efficacy studies. While MABs are a great tool for blocking specific targets, it should be noted that failure to inhibit one target may not apply to another. This is the case for IL13 and CD20. While CD20 blockade by rituximab in combination has been shown to be effective, results for rituximab alone under NCT01969409 are pending. However, IL13 inhibition by lebrikizumab was not effective in the phase 2 clinical trial under NCT01872689. Similar IL13 ineffectiveness results for tralokinumab are shown in Table 2.d under NCT01629667. LOXL2 was also evaluated as a potential target for IPF using MABs under simtuzumab in the phase 2 study under NCT01769196 and was also found ineffective.
- The review found that some classes of drugs are ineffective for IPF. This could be explained because they have little correlation with complex pathogenesis to drive previous efficacy studies. These classes include phosphodiesterase-5 (PDE-5) inhibitors and endothelin receptor antagonists (ERAs).
- PDE-5 inhibitors have been evaluated in several studies for IPF with sildenafil under NCTs NCT00517933, NCT02802345 and NCT00981747. Whether used as monotherapy or combination therapy, PDE-5 inhibitors have not been shown to be effective in IPF.
- Similarly, several members of ERAs were evaluated to determine if they would be beneficial for IPF in clinical trials under NCTs NCT00391443, NCT00768300 and NCT00903331. The same conclusion as for PDE-5 inhibitors could also be drawn for ERAs, as they are not effective in IPF.
Patients with IPF have been found to have lung dysbiosis, an imbalance between the types of organisms normally present in the lung. This manifests as an increased bacterial load and/or a loss of normal flora genera. This can lead to disease progression and mount a systemic and local immune response. This immune response can cause acute worsening of IPF which can lead to hospitalization and decrease in survival. Antibiotic therapy has been studied to show a favorable change in the flora in other chronic disorders. Initial randomized trials with co-trimoxazole (trimethoprim-sulfamethoxazole) in patients with IPF showed an improvement, however when compared with a large placebo-controlled trial, there was no significant improvement between the placebo and co-trimoxazole. In this trial, 513 individuals with IPF were given co-trimoxazole or doxycycline with the usual care. The addition of the antibiotics did not significantly improve the time to return from hospitalization or avoiding death. This study showed no benefit in treating the underlying disease with these antibiotics. 42 There have also been studies on the use of antivirals, as many individuals suffering from IPF have been found to have human herpes viruses, Epstein-Barr virus, and cytomegalovirus living in the lungs. A phase 1B randomized, prospective, placebo-controlled, double-blind clinical trial was completed to see how well patients tolerated the addition of the antiviral valganciclovir to the FDA-approved therapy (pirfenidone). Upon completion, the study showed the addition of valganciclovir was well tolerated and had no limiting safety signals, with data that showed preliminary efficacy. These results support the initiation of a larger trial to focus on efficacy.33
In the case of IPF, delivering an effective dose directly to the lung may be beneficial. There are four types of devices currently used for the administration of inhaled drugs: Pressurized metered dose inhalers (pMDI), Dry powder inhalers (DPI), Respimat soft mist inhalers (SMI), and nebulizers. Reports suggest that deposition is approximately twice as high using SMI as compared to a DPI formulation.[57] By delivering a drug directly to the site of action, efficacy can be improved. Side effects can be significantly reduced by reducing toxicity. Targets that are effective but have concerns about side effects may consider delivering the drug using a pulmonary drug delivery device.
Conclusions
Despite an exponential increase in knowledge and the emergence of novel therapies, approved treatments remain unsatisfactory for a significant proportion of patients. This is partly because the cause of the disease is complex, leading to multiple approaches being explored to see which would be effective as therapy targets. We conducted this review to determine the status of what therapies are in the pipeline for IPF. Based on the clinical studies, antifibrotic agents with specific targets appear to be the most promising. Some therapies that fall under this in terms of disease progression include anti-CTGF antibodies, LPA1 antagonists, recombinant human pentraxin-2 protein, and G protein-coupled receptor 84 antagonists. Because of the ECM factor of IPF, other potential therapies, such as reductive autoantibody therapy using anti-CD20 monoclonal antibodies, prostacyclin analogues, and stem cells, may have an advantage in modifying disease progression. Due to the complex pathophysiology of IPF, combination therapy with an anti-inflammatory agent may be considered. To optimize the treatment delivery of such promising therapies, a lung-specific delivery by inhalation that minimizes delivery to other sites in the body could limit the side effects observed in studies.
Acknowledgments
The authors are thankful Dr. Randy Mullins, Department Chair of Pharmaceutical Sciences, Dr. Angie Mutter, Director of Student Success and Career Planning, Ginny Patel, PharmD Candidate Class of 2023, and Trevor Hartman, PharmD Candidate Class of 2023 at Appalachian College of Pharmacy for their time and effort in reviewing the manuscript and providing input to complete this project.
References
- Valenzuela C, Torrisi SE, Kahn N, Quaresma M, Stowasser S, Kreuter M. Ongoing challenges in pulmonary fibrosis and insights from the nintedanib clinical programme. Respiratory Research 2020;21(1):1-15.
- George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. The Lancet Respiratory Medicine 2020;8(8):807-815.
- Foundation PF. Idiopathic Pulmonary Fibrosis
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. European Respiratory Review 2012;21(126):355-361.
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. European Respiratory Journal 2015;46(3):795-806.
- Barratt SL, Creamer A, Hayton C, Chaudhuri N. Idiopathic pulmonary fibrosis (IPF): an overview. Journal of clinical medicine 2018;7(8):201.
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet journal of rare diseases 2008;3(1):1-15.
- Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. New England Journal of Medicine 2018;378(19):1811-1823.
- Fraser E, Hoyles RK. Therapeutic advances in idiopathic pulmonary fibrosis. Clinical Medicine 2016;16(1):42.
- Waters DW, Blokland KE, Pathinayake PS, Burgess JK, Mutsaers SE, Prele CM, et al. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. American Journal of Physiology-Lung Cellular and Molecular Physiology 2018;315(2):L162-L172.
- O’Donoghue RJ, Knight DA, Richards CD, Prêle CM, Lau HL, Jarnicki AG, et al. Genetic partitioning of interleukin‐6 signalling in mice dissociates Stat3 from Smad3‐mediated lung fibrosis. EMBO molecular medicine 2012;4(9):939-951.
- White ES, Thomas M, Stowasser S, Tetzlaff K. Challenges for clinical drug development in pulmonary fibrosis. Frontiers in Pharmacology 2022;13:90.
- Ushakumary MG, Riccetti M, Perl A-KT. Resident interstitial lung fibroblasts and their role in alveolar stem cell niche development, homeostasis, injury, and regeneration. Stem cells translational medicine 2021;10(7):1021-1032.
- NIH. IDIOPATHIC PULMONARY FIBROSIS. Causes and Risk Factors
- Fujita Y. Extracellular vesicles in idiopathic pulmonary fibrosis: pathogenesis and therapeutics. Inflammation and Regeneration 2022;42(1):1-8.
- Richeldi L, Ryerson CJ, Lee JS, Wolters PJ, Koth LL, Ley B, et al. Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax 2012;67(5):407-411.
- Kreuter M, Del Galdo F, Miede C, Khanna D, Wuyts WA, Hummers LK, et al. Impact of lung function decline on time to hospitalisation events in systemic sclerosis-associated interstitial lung disease (SSc-ILD): a joint model analysis. Arthritis Research & Therapy 2022;24(1):1-9.
- Astrazeneca. US FDA grants saracatinib Orphan Drug Designation for idiopathic pulmonary fibrosis
- FDA U. Package Insert/OFEV® (nintedanib) capsules, for oral use
- FDA U. Package Insert/ESBRIET® (pirfenidone) capsules and film-coated tablets, for oral use
- NIH/Clinicaltrials.gov. Idiopathic Pulmonary Fibrosis
- Ng B, Dong J, D’Agostino G, Viswanathan S, Widjaja AA, Lim W-W, et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Science Translational Medicine 2019;11(511):eaaw1237.
- FDA U. The Voice of the Patient
- Yavari M, Mousavi SAJ, Janani L, Feizy Z, Vafa M. Effects of supplementation of vitamins D, C and E on Idiopathic Pulmonary Fibrosis (IPF): A clinical trial. Clinical Nutrition ESPEN 2022.
- Karampitsakos T, Vraka A, Bouros D, Liossis S-N, Tzouvelekis A. Biologic treatments in interstitial lung diseases. Frontiers in medicine 2019;6:41.
- Horton MR, Santopietro V, Mathew L, Horton KM, Polito AJ, Liu MC, et al. Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: a randomized trial. Annals of Internal Medicine 2012;157(6):398-406.
- Donahoe M, Valentine VG, Chien N, Gibson KF, Raval JS, Saul M, et al. Autoantibody-targeted treatments for acute exacerbations of idiopathic pulmonary fibrosis. PloS one 2015;10(6):e0127771.
- Richeldi L, Pérez ERF, Costabel U, Albera C, Lederer DJ, Flaherty KR, et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): a phase 2, randomised, double-blind, placebo-controlled trial. The Lancet Respiratory Medicine 2020;8(1):25-33.
- Raghu G, Van Den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. Jama 2018;319(22):2299-2307.
- Palmer SM, Snyder L, Todd JL, Soule B, Christian R, Anstrom K, et al. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest 2018;154(5):1061-1069.
- Cottin V, Seemayer CA, Fagard L, Ford P, Van Der Aa T, De Haas-Amatsaleh A, et al. Results of a phase 2 study of GLPG1205 for idiopathic pulmonary fibrosis (PINTA)
- Hirani N, MacKinnon AC, Nicol L, Ford P, Schambye H, Pedersen A, et al. Target inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. European Respiratory Journal 2021;57(5).
- Blackwell TS, Hewlett JC, Mason WR, Martin S, Del Greco J, Ding G, et al. A phase I randomized, controlled, clinical trial of valganciclovir in idiopathic pulmonary fibrosis. Annals of the American Thoracic Society 2021;18(8):1291-1297.
- Chambers D, Enever D, Ilic N, Sparks L, Ayres J, Yerkovich S, et al. A phase 1b study of mesenchymal stromal cell therapy for idiopathic pulmonary fibrosis. Cytotherapy 2014;16(4):S12.
- Network IPFCR. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. New England Journal of Medicine 2012;366(21):1968-1977.
- Behr J, Bendstrup E, Crestani B, Günther A, Olschewski H, Sköld CM, et al. Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: a randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Respiratory Medicine 2016;4(6):445-453.
- Raghu G, Richeldi L, Crestani B, Wung P, Bejuit R, Esperet C, et al. SAR156597 in idiopathic pulmonary fibrosis: a phase 2 placebo-controlled study (DRI11772). European Respiratory Journal 2018;52(6).
- Raghu G, Mouded M, Chambers DC, Martinez FJ, Richeldi L, Lancaster LH, et al. A phase IIb randomized clinical study of an anti-αvβ6 monoclonal antibody in idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine 2022;206(9):1128-1139.
- Parker JM, Glaspole IN, Lancaster LH, Haddad TJ, She D, Roseti SL, et al. A phase 2 randomized controlled study of tralokinumab in subjects with idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine 2018;197(1):94-103.
- Raghu G, Brown KK, Collard HR, Cottin V, Gibson KF, Kaner RJ, et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, phase 2 trial. The Lancet Respiratory Medicine 2017;5(1):22-32.
- Maher TM, Costabel U, Glassberg MK, Kondoh Y, Ogura T, Scholand MB, et al. Phase 2 trial to assess lebrikizumab in patients with idiopathic pulmonary fibrosis. European Respiratory Journal 2021;57(2).
- Martinez FJ, Yow E, Flaherty KR, Snyder LD, Durheim MT, Wisniewski SR, et al. Effect of antimicrobial therapy on respiratory hospitalization or death in adults with idiopathic pulmonary fibrosis: the CleanUP-IPF randomized clinical trial. Jama 2021;325(18):1841-1851.
- Rosas IO, Goldberg HJ, Collard HR, El-Chemaly S, Flaherty K, Hunninghake GM, et al. A phase II clinical trial of low-dose inhaled carbon monoxide in idiopathic pulmonary fibrosis. Chest 2018;153(1):94-104.
- Network IPFCR. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. New England Journal of Medicine 2010;363(7):620-628.
- Behr J, Kolb M, Song JW, Luppi F, Schinzel B, Stowasser S, et al. Nintedanib and sildenafil in patients with idiopathic pulmonary fibrosis and right heart dysfunction. A prespecified subgroup analysis of a double-blind randomized clinical trial (INSTAGE). American Journal of Respiratory and Critical Care Medicine 2019;200(12):1505-1512.
- King Jr TE, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine 2011;184(1):92-99.
- Kale A, Sharma A, Stolzing A, Desprez P-Y, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immunity & Ageing 2020;17(1):1-9.
- Nathan SD, Behr J, Cottin V, Lancaster L, Smith P, Deng C, et al. Study design and rationale for the TETON phase 3, randomised, controlled clinical trials of inhaled treprostinil in the treatment of idiopathic pulmonary fibrosis. BMJ Open Respiratory Research 2022;9(1):e001310.
- Ruigrok MJ, Frijlink HW, Melgert BN, Olinga P, Hinrichs WL. Gene therapy strategies for idiopathic pulmonary fibrosis: recent advances, current challenges, and future directions. Molecular Therapy-Methods & Clinical Development 2021;20:483-496.
- NIH/Clinicaltrials.gov. Autoantibody Reduction Therapy in Patients With Idiopathic Pulmonary Fibrosis (ART-IPF)
- Averyanov A, Koroleva I, Konoplyannikov M, Revkova V, Lesnyak V, Kalsin V, et al. First-in-human high-cumulative-dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem cells translational medicine 2020;9(1):6-16.
IPF Pipeline: What is the Current and Future Status on Idiopathic Pulmonary Fibrosis Drug Development? © 2024 by Dalton P. Oliver, Mohammad Faisal Hossain is licensed under CC BY 4.0![]()
![]()
Note
Copyright © 2024 Oliver et al. Place of Publication: PSciP Publishing LLC, Oakwood, VA, USA.